Lab 3:
Polymerase Chain Reaction (PCR)
PRE-LAB CONCEPTS
Introduction
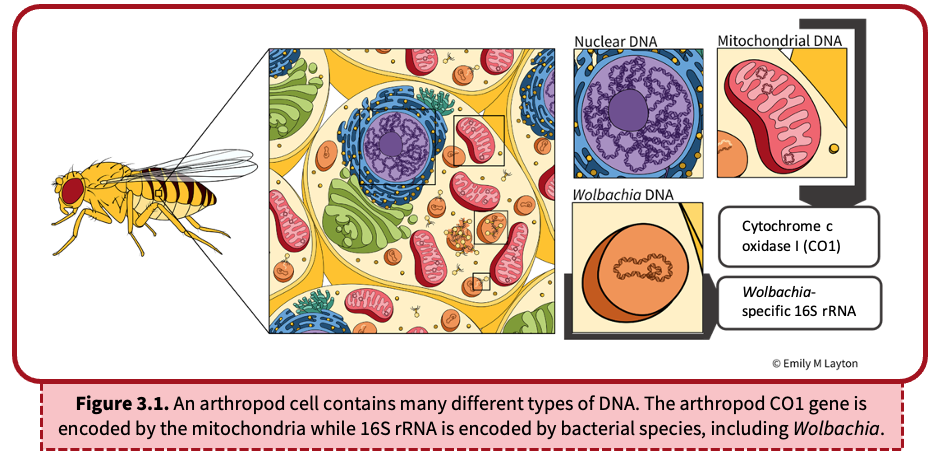
DNA can be found in many different places in the arthropod (Figure 3.1). The DNA extraction process (Lab 2) purified all DNA from the sample including nuclear and mitochondrial DNA of the arthropod, as well as bacterial DNA. In this activity, we will use Polymerase Chain Reaction (PCR) to amplify segments of the extracted DNA in order to (i) obtain enough DNA for arthropod identification and (ii) determine whether or not the arthropod is infected with Wolbachia. We will do this by targeting two specific genes: CO1 from the arthropod and 16S rRNA from Wolbachia.
Arthropod CO1
Cytochrome c oxidase I (CO1) is a component of mitochondria, energy-producing organelles within the cells of most eukaryotes (Figure 3.1). Because a single animal cell can contain hundreds to thousands of mitochondria, and each mitochondrion encodes multiple copies of mitochondrial DNA (mtDNA), the mitochondrial CO1 gene is an excellent candidate for PCR amplification. The DNA sequence of the CO1 gene is unique to each species and serves as a useful tool to identify organisms, termed barcoding. This is particularly helpful when classifying closely related arthropods that are often difficult to differentiate by eye.
Wolbachia-specific 16S rRNA
16S rRNA, a component of the prokaryotic 30S ribosomal subunit, is the most commonly used gene for bacterial detection (Figure 3.1). Similar to CO1, it contains a unique DNA sequence that allows for general detection by PCR and can facilitate species identification. For the purpose of this lab, we will use a PCR assay that specifically targets Wolbachia 16S rRNA. If Wolbachia is present in the cell, the Wolbachia 16S rRNA gene will be amplified. If absent, the Wolbachia-specific DNA sequence will not be amplified. Likewise, other non-Wolbachia bacteria will not be amplified. We will visualize the presence/absence of this DNA amplification using gel electrophoresis (see Lab 4). Finally, we can also use DNA sequencing to identify specific strains of Wolbachia and infer evolutionary relationships among closely related Wolbachia supergroups.
DNA Barcoding
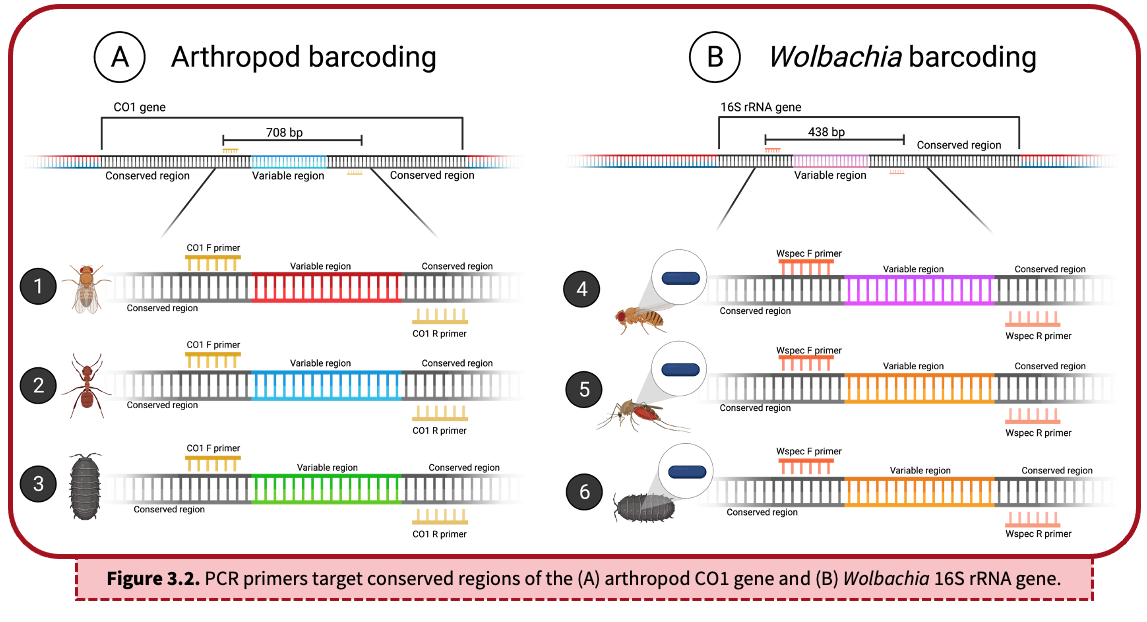
A DNA barcode refers to a unique sequence of DNA that can be used to identify organisms. This barcode sequence is compared to a collection of other known DNA sequences, termed a reference database, to find the best match. There are two key components of a DNA barcode sequence:
- Conserved region: Shown in black, the conserved region is a sequence of DNA that is the same across a group of organisms – in this case, arthropods (A) or Wolbachia (B). Due to sequence conservation of this region, we are able to target and amplify specific DNA using PCR primers. The same set of PCR primers will amplify DNA from most arthropods, including flies, ants, and pillbugs (as shown above). A second set of PCR primers will amplify DNA from Wolbachia.
- Variable region: An internal segment of unique DNA is sequenced in order to taxonomically classify species. As seen in (A), each organism encodes a unique, internal variable region (illustrated in red, blue, and green) because they represent three different arthropods. In (B), the mosquito and pillbug (#5 & 6) share a similar 16S rRNA region (shown in orange) because they are infected with similar Wolbachia The fruit fly (#4), however, is infected with a different Wolbachia strain and therefore has a unique variable region (shown in pink). It is important to note that ALL sequences above will be amplified with this PCR lab because we are targeting the conserved regions of the CO1 and 16S rRNA genes.
The following three sequences below represent a small segment of the Wolbachia 16S rRNA variable region. Once the region is amplified with PCR, we can sequence the DNA in order to “barcode” the organism. Evolutionary relatedness is determined by identity (i.e., nucleotides that are the same) across the sequences. Sequences 2 and 3 share 100% identity across the 30-nucleotide region and represent similar strains of Wolbachia. Sequence 1, however, represents a different Wolbachia strain based on nucleotide variation at positions 1,3, and 29. Based on this short region, Sequence 1 shares 90% identity (27/30 bases) with the other two DNA sequences.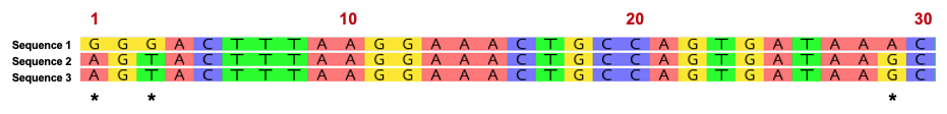
Polymerase Chain Reaction (PCR)
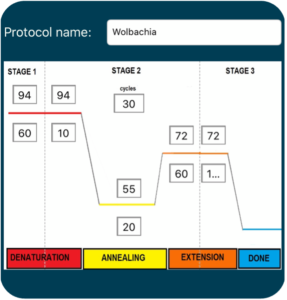
Polymerase Chain Reaction (PCR) is a common laboratory technique used to amplify DNA. The small amounts of DNA that are obtained using DNA extraction are generally not enough to visualize and thoroughly analyze, so PCR is necessary to exponentially amplify DNA of interest. A single PCR cycle consists of three distinct steps – denaturation, annealing, and extension (Figure 3.3) – and this cycle is repeated several times. After each cycle of PCR, the amount of DNA is doubled. Assuming we only start with a single molecule of DNA, 30 cycles of PCR would yield over a billion copies of the target DNA.
Denaturation
Denaturation is the first step of the PCR process. In this step, DNA is heated to a high temperature (typically 92-94°C) to unwind and separate the double stranded DNA molecules into two complementary single strands.
Annealing
The second stage of the PCR process is called annealing. During this stage, the reaction temperature is lowered to facilitate binding of PCR primers to the denatured, single-stranded DNA. Specific annealing temperature varies depending on the sequence and length of the primers. Forward and reverse PCR primers are small strands of nucleotides that are designed to target and amplify specific portions of DNA. They bind to each end of the denatured DNA strand based off of complementary base pairing rules (i.e., A-T and C-G) and serve as the scaffold for a new complementary strand of DNA.
Extension
The final stage of PCR is an extension phase where the temperature is slightly raised so the enzyme Taq polymerase can add complementary nucleotides to the template strand beginning at the location of the primer. With the primer serving as the scaffold of the complementary strand, the primer is extended to generate a double stranded DNA molecule that is identical to the original template. Once extension is complete, the temperature is raised again to begin the denaturation step of the next cycle.

Key Elements for PCR
DNA
DNA from the arthropod DNA extraction (Lab 2) will be amplified during PCR. Heat is used to unwind the double stranded DNA, resulting in two complementary single strands. The two single strands now act as templates to generate new double stranded molecules of DNA.
Nucleotides
Nucleotides, also called dNTPs (deoxynucleotide triphosphates), bases or DNA bases, are single units of Adenine (A), Thymine (T), Cytosine (C), and Guanine (G). They must be added to the PCR reaction and serve as building blocks for new DNA molecules.
Primers
Primers are small lengths of DNA, generally around 20 nucleotides, that are designed to bind and amplify a specific section or gene of the DNA strand. They are needed because DNA polymerase, the enzyme that adds nucleotides to the single stranded DNA, can only add to an existing nucleotide. Because the primers must be specific to the strand, abiding by the typical base pairing rules, you generally need to know the DNA sequence that you wish to amplify before you begin the PCR process.
Buffer
A buffer will be added to the PCR mix in order to maintain pH conditions for the entirety of the reaction and promote primer binding.
Taq polymerase
This enzyme acts as a DNA polymerase to add new DNA bases to the end of the primer sequence using the base pairing rules of nucleotides. PCR was made possible through the discovery of this thermostable enzyme in Thermus aquaticus, an extremophile isolated from the Lower Geyser Basin of Yellowstone National Park. While most DNA polymerases are temperature sensitive, Taq is able to withstand the high temperatures needed to denature DNA in PCR and is thus used as the primary source of extension in modern PCR reactions.

Note: For ease and optimization of PCR, many companies sell a Taq Master Mix consisting of Taq polymerase, nucleotides, and buffer. Therefore, only primers and DNA need to be added to the mix. Water may be added to bring the reaction to the desired volume.
RECOMMENDED LAB GUIDE
Why do we recommend this protocol?
This lab activity involves two separate PCR reactions (Arthropod and Wolbachia) which allows for customization of the annealing temperature. The protocol is recommended for more sensitive Wolbachia detection and downstream analyses, such as Sanger sequencing.
Relative to the duplex reaction:
- Pros: Better detection of Wolbachia and better visualization of bands in Lab 4.
- Cons: Requires more consumable materials (for PCR and gel electrophoresis) and additional class time.
Standard PCR Protocol
The protocol below has been enhanced with interactive Discovery Tools. Click on the highlighted text preceding the Discovery icon ![]() to view supporting media.
to view supporting media.














Prepare the Thermal Cycler
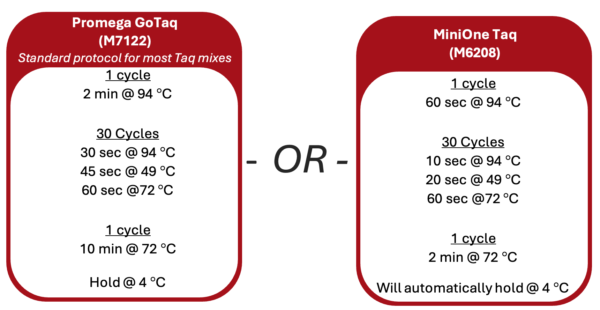
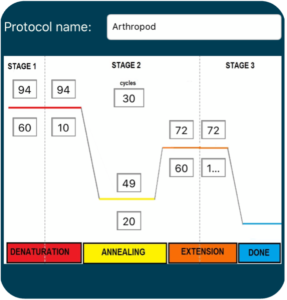
1. Turn on the thermal cycler and enter the appropriate CO1 PCR program ![]() to accommodate the brand of Taq polymerase.
to accommodate the brand of Taq polymerase.
2. If using a MiniOne thermal cycler, refer to the Getting Started Guide to set up the program.
Prepare Lab Space and Label Tubes
3. Remove all unnecessary items from your lab station.
4. Put on nitrile gloves and clean all surfaces by wiping down with 70% Ethanol.
5. Collect a 1.5 mL microcentrifuge tube ![]() . Label the tube “A” (for Arthropod PCR Cocktail). Place the tube in a 1.5mL tube rack.
. Label the tube “A” (for Arthropod PCR Cocktail). Place the tube in a 1.5mL tube rack.
6. Collect six 0.2 mL PCR tubes ![]() . Number and label them with your initials. Place the tubes in a PCR tube rack and record putative identification of each arthropod in the table.
. Number and label them with your initials. Place the tubes in a PCR tube rack and record putative identification of each arthropod in the table.
| Label | Contents |
|---|---|
| (Initials) 1 | Arthropod #1 ID: __________ |
| (Initials) 2 | Arthropod #2 ID: __________ |
| (Initials) 3 | Positive (+) Arthropod control |
| (Initials) 4 | Negative (-) Arthropod control |
| (Initials) 5 | Positive (+) DNA control |
| (Initials) 6 | Water control |
Add Template DNA to PCR Tubes
7. Use a P-2 or P-20 pipette to add 2 μL of template DNA ![]() to each corresponding Arthropod PCR tube above. It is critical that you change tips between each tube.
to each corresponding Arthropod PCR tube above. It is critical that you change tips between each tube.
8. Set aside the PCR tubes for now. Place the template DNA tubes back into storage. You will need these again for the Wolbachia PCR.
Prepare PCR Cocktail
9. Use a P-200 pipette to add each of the following reagents ![]() (from the “Total for 7 reactions” column) to the 1.5 mL tube marked “A”. Change tips between each reagent, and check off each reagent after it is added.
(from the “Total for 7 reactions” column) to the 1.5 mL tube marked “A”. Change tips between each reagent, and check off each reagent after it is added.
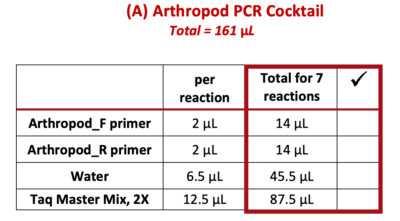
Note: PCR best practice is to make a PCR Cocktail for the (number of samples) + 1. This will account for any liquid lost due to retention in the pipette tip or from small air bubbles in the reaction. In this case, we will make a PCR Cocktail for 7 reactions (6 samples + 1).
10. Close the tube and briefly vortex ![]() for 5 seconds.
for 5 seconds.
11. Place the A tube on one side of the mini-centrifuge and the B (balancer tube) on the opposite side. This is called “balancing” the rotor; if the tubes are not balanced, it will make a loud sound and could damage the microcentrifuge. Quickly spin down ![]() (~ 3-5 seconds) the Arthropod PCR Cocktail to collect liquid at the bottom of the tube.
(~ 3-5 seconds) the Arthropod PCR Cocktail to collect liquid at the bottom of the tube.
Set Up PCR Reaction
 12. Use a P-200 pipette to add 23 µL of the Arthropod PCR Cocktail (A) to tubes 1-6
12. Use a P-200 pipette to add 23 µL of the Arthropod PCR Cocktail (A) to tubes 1-6 ![]() (which should already contain the template DNA). Change tips between each tube. Never place a used pipette tip into the PCR Cocktail because it will contaminate all downstream reactions.
(which should already contain the template DNA). Change tips between each tube. Never place a used pipette tip into the PCR Cocktail because it will contaminate all downstream reactions.
13. Tightly secure the lids on each tube.
14. Change the rotor ![]() on your mini-centrifuge to the PCR tube rotor. Center tubes 1-3 on one side and 4-6 on the other side to balance the rotor. Briefly spin
on your mini-centrifuge to the PCR tube rotor. Center tubes 1-3 on one side and 4-6 on the other side to balance the rotor. Briefly spin ![]() to collect all liquid at the bottom of the tubes.
to collect all liquid at the bottom of the tubes.
15. Transfer the tubes to the thermal cycler. Once everyone has placed their samples in the thermocycler, the program can be started ![]() with the Arthropod PCR protocol. If using MiniOne, you may observe the PCR program as it cycles.
with the Arthropod PCR protocol. If using MiniOne, you may observe the PCR program as it cycles.
16. Clean up your lab station and wipe surfaces with ethanol.
Storage
17. When the thermal cycler is done, store samples in the refrigerator (4°C).
18. Proceed to the Wolbachia PCR reaction.
Prepare the Thermal Cycler
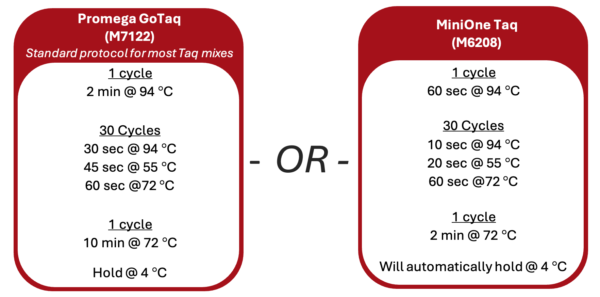
1. Turn on the thermal cycler and enter the appropriate Wolbachia PCR program following the guide below.
2. If using a MiniOne thermal cycler, refer to the Getting Started Guide to set up the program.
Prepare Lab Space and Label Tubes
3. Remove all unnecessary items from your lab station.
4. Put on nitrile gloves and clean all surfaces by wiping down with 70% Ethanol.
5. Collect a 1.5 mL microcentrifuge tube ![]() . Label the tube “W” (for Wolbachia PCR Cocktail). Place the tube in a 1.5mL tube rack.
. Label the tube “W” (for Wolbachia PCR Cocktail). Place the tube in a 1.5mL tube rack.
6. Collect six 0.2 mL PCR tubes ![]() . Number and label them with your initials. Place the tubes in a PCR tube rack and record putative identification of each arthropod in the table.
. Number and label them with your initials. Place the tubes in a PCR tube rack and record putative identification of each arthropod in the table.
| Label | Contents |
|---|---|
| (Initials) 1 | Arthropod #1 ID: __________ |
| (Initials) 2 | Arthropod #2 ID: __________ |
| (Initials) 3 | Positive (+) Arthropod control |
| (Initials) 4 | Negative (-) Arthropod control |
| (Initials) 5 | Positive (+) DNA control |
| (Initials) 6 | Water control |
Add Template DNA to PCR Tubes
7. Use a P-2 or P-20 pipette to add 2 μL of template DNA ![]() to each corresponding Arthropod PCR tube above. It is critical that you change tips between each tube.
to each corresponding Arthropod PCR tube above. It is critical that you change tips between each tube.
8. Set aside the PCR tubes for now. Place the template DNA tubes back into storage.
Prepare PCR Cocktail
9. Use a P-200 pipette to add each of the following reagents ![]() (from the “Total for 7 reactions” column) to the 1.5 mL tube marked “W”. Change tips between each reagent, and check off each reagent after it is added.
(from the “Total for 7 reactions” column) to the 1.5 mL tube marked “W”. Change tips between each reagent, and check off each reagent after it is added.
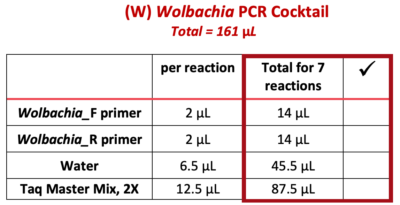
Note: PCR best practice is to make a PCR Cocktail for the (number of samples) + 1. This will account for any liquid lost due to retention in the pipette tip or from small air bubbles in the reaction. In this case, we will make a PCR Cocktail for 7 reactions (6 samples + 1).
10. Close the tube and briefly vortex ![]() for 5 seconds.
for 5 seconds.
11. Place the W tube on one side of the mini-centrifuge and the B (balancer tube) on the opposite side. This is called “balancing” the rotor; if the tubes are not balanced, it will make a loud sound and could damage the microcentrifuge. Quickly spin down ![]() (~ 3-5 seconds) the Wolbachia PCR Cocktail to collect liquid at the bottom of the tube.
(~ 3-5 seconds) the Wolbachia PCR Cocktail to collect liquid at the bottom of the tube.
Set Up PCR Reaction
 12. Use a P-200 pipette to add 23 µL of the Wolbachia PCR Cocktail (W) to tubes 1-6
12. Use a P-200 pipette to add 23 µL of the Wolbachia PCR Cocktail (W) to tubes 1-6 ![]() (which should already contain the template DNA). Change tips between each tube. Never place a used pipette tip into the PCR Cocktail because it will contaminate all downstream reactions.
(which should already contain the template DNA). Change tips between each tube. Never place a used pipette tip into the PCR Cocktail because it will contaminate all downstream reactions.
13. Tightly secure the lids on each tube.
14. Change the rotor ![]() on your mini-centrifuge to the PCR tube rotor. Center tubes 1-3 on one side and 4-6 on the other side to balance the rotor. Briefly spin
on your mini-centrifuge to the PCR tube rotor. Center tubes 1-3 on one side and 4-6 on the other side to balance the rotor. Briefly spin ![]() to collect all liquid at the bottom of the tubes.
to collect all liquid at the bottom of the tubes.
15. Transfer the tubes to the thermal cycler. Once everyone has placed their samples in the thermocycler, the program can be started ![]() with the Wolbachia PCR protocol. If using MiniOne, you may observe the PCR program as it cycles.
with the Wolbachia PCR protocol. If using MiniOne, you may observe the PCR program as it cycles.
16. Clean up your lab station and wipe surfaces with ethanol.
Storage
17. When the thermal cycler is done, store samples in the refrigerator (4°C).
18. Proceed to Lab 4: Gel Electrophoresis.
- Thermal cycler
 (PCR machine)
(PCR machine) - Vortex Mixer
- Mini-centrifuge
- Nitrile gloves
- Squirt bottle or spray bottle with 70% ethanol
- 0.2 mL PCR tubes
- Rack for 0.2 mL PCR tubes
- 1.5 mL tubes
- Rack for 1.5 mL tubes
- Balance tube
- Waste cup for tips
- Micropipettes (20 μL and 200 μL)
- Micropipette tips (20 μL and 200 μL )
- Sharpie
- 2 DNA samples from arthropod specimens (Lab 2)
- +/- DNA from arthropod controls (Lab 2)
- + DNA control
- Sterile, nuclease-free water
- Taq Master Mix
- Wolbachia_F and Wolbachia_R PCR primers
- Arthropod_F and Arthropod_R primers
Introduction
In this lab, you will learn what Polymerase Chain Reaction (PCR) does, how it works, and why it is useful to research in the biological sciences. You will use PCR to amplify any Wolbachia 16S rRNA (if present) and amplify the arthropod CO1 (barcoding) gene from the extracted DNA of the two selected specimens and control insects. You will also amplify a previously extracted DNA sample which is known to be positive for Wolbachia (positive control) and a water sample (negative control).
Primers
There are two sets of primers used to amplify fragments of interest, labelled “Wolbachia_F/Wolbachia_R” and “Arthropod_F/Arthropod_R”. Primers always come in sets, with a forward and reverse direction. It is vital to add both forward and reverse primers to the PCR mix. The original names for each primer set are included in parentheses below.
Primers to specifically amplify a 438-bp (base pair) fragment of the 16S ribosomal RNA gene (ubiquitous in all Wolbachia) are:
Wolbachia_F (16S_WspecF): 5’−CAT ACC TAT TCG AAG GGA TAG−3’
Wolbachia_R (16S_WspecR): 5’−AGC TTC GAG TGA AAC CAA TTC−3’
When using these primers, the ideal annealing temperature is 55°C.
Primers to specifically amplify a 708-bp fragment of the CO1 cytochrome oxidase gene (ubiquitous in arthropod mitochondria) are:
Arthropod_F (LCO1490): 5’−GGT CAA CAA ATC ATA AAG ATA TTG G−3’
Arthropod_R (HCO2198): 5’−TAA ACT TCA GGG TGA CCA AAA AAT CA−3’
When using these primers, the ideal annealing temperature is 49°C.
PCR Mixes
Different ingredients are needed for a PCR. These ingredients are added in different steps for clarity. The protocol refers to two different mixes:
- Taq Master Mix – this mix will be supplied directly from a vendor and includes Taq polymerase, dNTPs, and buffer.
- PCR Cocktail – you will create this mix by combining the Taq Master Mix with forward primer, reverse primer, and water.
Controls
Controls are used to minimize all variables except for the independent variables being tested; they are essential to ensuring the quality of the experiment. A positive control consists of a well-understood variable and is designed to produce an expected result. A negative control ensures that the samples and process are not contaminated; it is designed to produce a negative result.
This lab series features two sets of experimental controls:
- DNA Extraction Controls
-
(+) Control: An arthropod that is infected with Wolbachia and known to contain both the arthropod barcoding gene CO1 and the Wolbachia 16S rRNA gene.
-
(-) Control: An uninfected arthropod that contains only the arthropod barcoding gene CO1.
-
- PCR Controls
-
(+) Control: DNA extracted from a Wolbachia-infected arthropod and verified to amplify both CO1 and Wolbachia 16S rRNA fragments.
-
(-) Control: Purified water is added to the PCR reaction instead of DNA. It should not produce any results because there is no template DNA.
-
Helpful Tips
To ensure optimal results:
- Avoid contamination by changing tips between each reagent/ sample.
- Avoid contamination by keeping the lab station free of clutter.
- Make sure you thoroughly thaw primers, water, and master mixes before using them.
- To save time, turn on and program the thermocycler before starting to prep samples. If using the MiniOne thermocycler you will need to download the app from their website or the Apple store to your phone or tablet (only one phone per thermocycler).
ALTERNATIVE PROTOCOL: DUPLEX PCR
What is duplex PCR?
This lab activity condenses the PCR protocol into one duplex reaction that amplifies both arthropod and Wolbachia DNA in the same assay. It will amplify most, but not all, Wolbachia as the PCR annealing temperature is optimized for the arthropod DNA. The protocol is recommended for efficient use of classroom time and resources.
FAQs
