Lab 4:
Gel electrophoresis
PRE-LAB CONCEPTS
Introduction
This lab will determine the presence or absence of amplified DNA in your samples by visualization on an agarose gel. Arthropod and Wolbachia DNA, if present, will be distinguishable based on the size, or base pair (bp) length, of the DNA molecule.
Gel electrophoresis
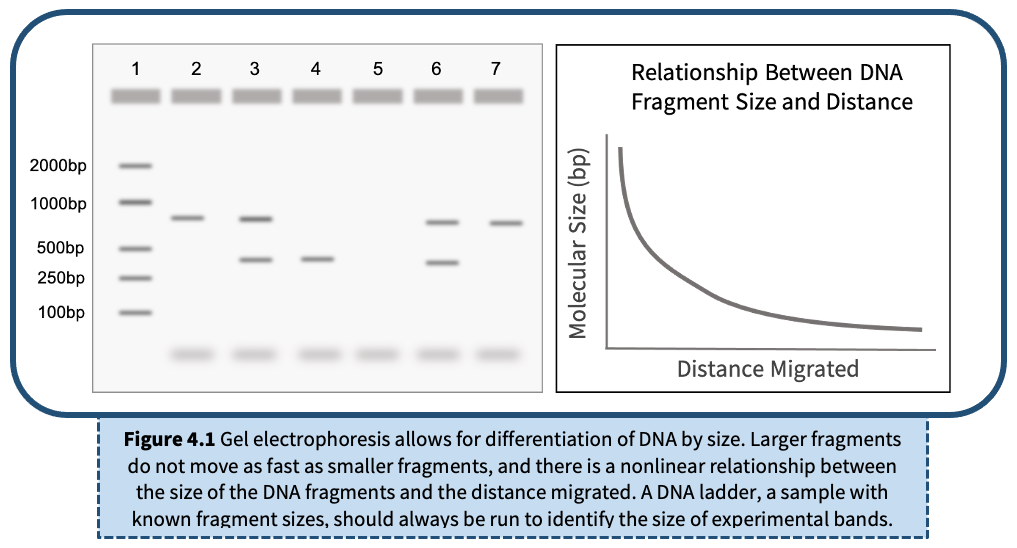
Gel electrophoresis is a method of separating DNA fragments by movement through a Jello-like substance called agarose. Derived from a seaweed polysaccharide, agarose gels form small pores that act as sieves to separate DNA based on size; whereby smaller DNA molecules move through the pores faster and easier than larger molecules. Loading wells are oriented at the top of the gel to allow for precise insertion of PCR products into the gel. An electrical current is applied to move negatively charged DNA molecules away from a negative electrode (-) and toward a positive electrode (+). DNA migrates through the gel in a single, vertical lane. Three factors influence the speed of movement: (i) the voltage of the electrical field, (ii) the concentration of agarose, and (iii) most importantly, the size of the DNA molecule.
DNA Visualization
DNA itself is not visible within an agarose gel. Therefore, a fluorescent stain is added to the gel that binds DNA and fluoresces under UV or blue light. DNA will appear as a horizontal line, or band, on the agarose gel.
Key Elements for Gel Electrophoresis
PCR Products (DNA)
The purpose of this lab is to visualize the PCR products, or amplified DNA, from your arthropod samples.
DNA Ladder
A DNA ladder is a cocktail of DNA fragments with pre-determined sizes. The ladder, also called a DNA marker, is loaded alongside experimental samples as a reference tool for estimating band size.
Agarose Gel
MiniOne GelCups contain solidified chunks of agarose that are melted and re-solidified in a casting tray to form the agarose gel. DNA will migrate through the gel and form separate bands based on size (correlating to length in bp).
DNA Stain
A DNA stain is added to the agarose gel to visualize DNA under a UV or blue light. It has already been pre-mixed into the MiniOne GreenGel GelCups.
Running Buffer
Running buffer is a conductive liquid that allows the DNA to migrate through the agarose gel. It is important that the agarose gel be made using the same buffer.
Loading Dye
Loading dye has two primary components: (i) a visible dye indicates how far the DNA has run on the gel and (ii) glycerol, which is denser than the buffer, ensures that samples fall into the loading wells rather than float back out. Some Taq Master Mixes (e.g., Promega GoTaq) already contain a pre-mixed loading dye.
Electrophoresis System
Running buffer and the agarose gel will be placed into the chamber of an electrophoresis system. After loading the samples, an electric current is applied to move the negatively charged DNA towards the positive end of the system. Without this electric field, the DNA will not migrate through the agarose gel. If the electric field is reversed, the DNA will run in the opposite direction, towards the top of the gel, and eventually exit the gel.
How to Read a Gel
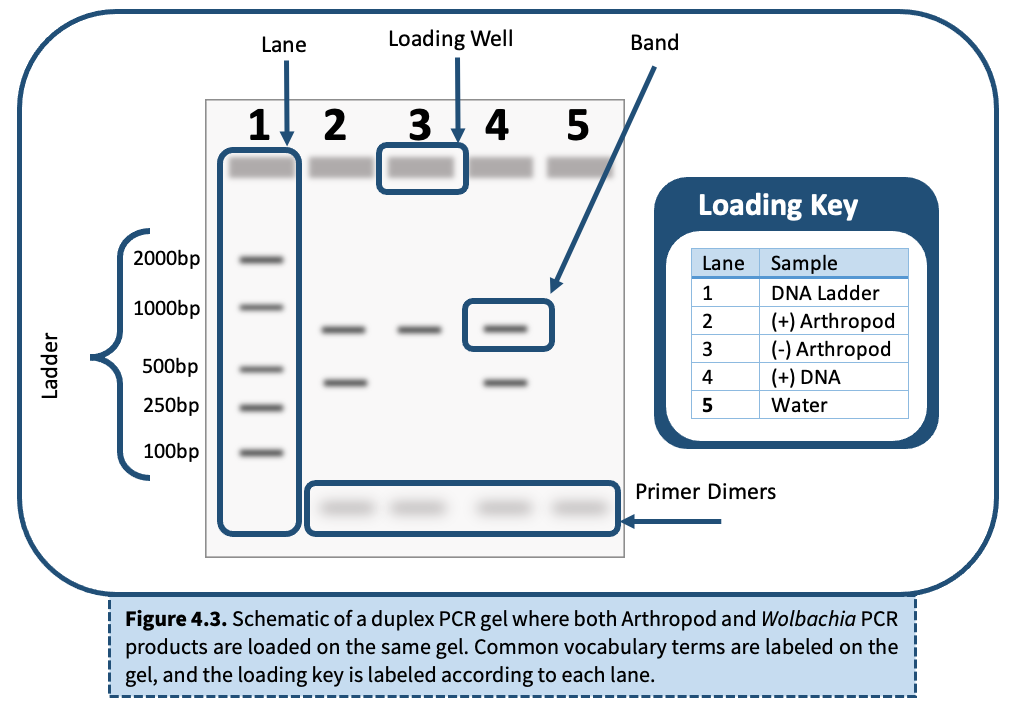
Lanes
DNA that was loaded into each well will migrate in a single, vertical lane towards the (+) charge.
Bands
When DNA becomes separated by size due to gel electrophoresis, they appear as bands in the gel. These are clearly defined, bright lines in the gel.
DNA Ladder
The DNA ladder will contain multiple bands in one lane. Each band represents a pre-determined length of DNA and can be used as a reference tool to estimate DNA size for each of the experimental samples. Refer to the product information for specific band sizes.
Primer Dimers
PCR reactions are set up with an excess of primers. In addition, some primers bind to each other instead of binding to the DNA, creating primer dimers. Primers are ~25bp long, so excess primers appear as fuzzy bands on the bottom of the gel ~25-50bp. This is normal and to be expected.
Reading a Single PCR vs. Duplex PCR Gel
A single PCR gel will contain only one amplified PCR product, either Wolbachia or arthropod, in each lane. A separate gel will need to be run for each DNA type.
A duplex PCR means that both the arthropod barcoding gene and the 16S rRNA fragment from Wolbachia were amplified in the same PCR reaction. When visualizing this PCR reaction, two bands should appear in the same lane if Wolbachia is present, and only one band will appear if the arthropod is uninfected.

How to Interpret Gel Electrophoresis Results
To interpret gel electrophoresis results, first ensure that all controls are correct. The DNA ladder, (+) Arthropod control, (-) Arthropod control, and (+) DNA control should produce bands of expected size, whereas the water lane should be empty.
- DNA Ladder
To accurately read the gel, confirm the band size of experimental samples by comparing their location in the gel to reference bands in the DNA Ladder. Refer to the information sheet accompanying your DNA ladder for specific band sizes as the bands, or rungs, vary by product. Below is the DNA Marker (M3104) from MiniOne.
- Positive Controls
The (+) Arthropod control and (+) DNA control should have both the CO1 and Wolbachia band present. In a duplex PCR, as shown below, these will appear in the same lane. In a standard (single) PCR, these will be loaded into separate gels. The DNA ladder bands should be clearly present and separated.
- Negative Controls
The (-) Arthropod control should have a CO1 band, but no Wolbachia band. The negative water control should not have any band or smudge.
If all controls worked, the results of your experiment are valid, and the experimental bands can be analyzed. If the controls have unexpected results, or if there is a band in the water lane, refer to the Troubleshooting section of the Lab Guide.
 .
.
RECOMMENDED LAB GUIDE
Why do we recommend this protocol?
The MiniOne System utilizes a built-in blue light and photo hood to allow real-time visualization of the bands as they move down the gel. Gels can be poured, run, and visualized in a single class period. For best results, we recommend using the MiniOne GreenGel GelCups (1% TBE).
The MiniPCR System uses a similar technology to the above protocol; refer to the manufacturer guide to adjust for slight differences with the equipment.
ALTERNATIVE PROTOCOL
This protocol can be used for most standard gel rigs. It will take generally take one class period to prepare and cast the gel, and a second class period to run and visualize the gel. Note that GelRed (or GelGreen) is added to the gel before it is cast. If you prefer post-staining the gel, you will need to allow an extra 30 min after the gel run to stain and image the gel. Refer to manufacturer guide for specific instructions. Most important: make sure the DNA Stain is compatible with your transillumator (blue light or UV).
ADDITIONAL RESOURCES
FAQs
